About
Documentation on metagenomes, reference genes, and annotation pipeline
Overview:
Microbiome researchers frequently want to know the abundance of a particular microbial gene, pathway, or species across different human hosts and its association with disease. While there are now thousands of publicly available metagenomes from the human gut, computational barriers prevent most researchers from conducting such analyses. MetaQuery is a web application that enables rapid and quantitative analysis of specific genes, functions and taxa across 1,267 publicly available human gut metagenomes. These data span several continents (Europe, China, North America) and disease states (IBD, diabetes, obesity, rheumatoid arthritis, colorectal cancer, and liver cirrhosis). The speed and accessibility of MetaQuery are a step toward democratizing metagenomics research, which should allow many researchers to query the abundance and variation of specific genes in the human gut microbiome.You can read more about MetaQuery here:
Nayfach S, Fischbach MA, Pollard KS. MetaQuery: a web server for rapid annotation and quantitative analysis of specific genes in the human gut microbiome. Bioinformatics 2015;31(14). doi:10.1093/bioinformatics/btv382
MetaQuery web development team at Gladstone Institutes:
Chunyu Zhao, Stephen Nayfach, Ayushi Agrawal, Andrew Davis, Alexander R. Pico, Katherine S. Pollard
How it works:
- Align query genes to the microbiome gene catalog: Query genes specified by the user are aligned to the integrated catalog of reference genes in the human gut microbiome using BLAST. More details about the gene catalog are provided below.
- Identify homologous microbiome genes: Gene sequences similar to any of the user's queries are identified in the gene catalog. Based on specified alignment parameters, the user can choose to target genes that are highly similar (e.g. >95% identity) or more distantly related (e.g. >30% identity).
- Quantify abundance of homologs: The abundance of all 9.9 million microbiome genes is precomputed for 1,267 samples. This enables MetaQuery to rapidly look up gene abundances for identified homologs. Gene abundances are defined as the average gene copies per cell. Read below for more information about how gene abundances are estimated.
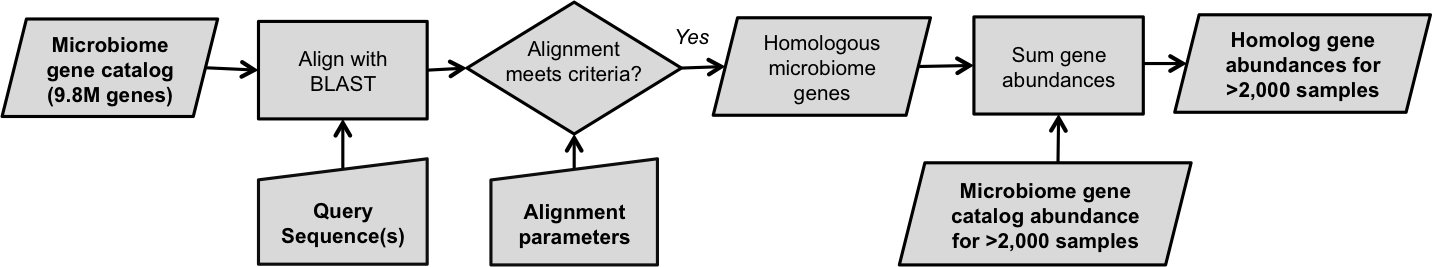
Flowchart for estimating the abundance a query gene in the human gut.
Microbiome gene catalog:
| count genes in catalog | 9879900.0 |
| % complete genes | 57.74 |
| % from reference genomes | 2.46 |
| % annotated at phylum level | 21.31 |
| % annotated at genus level | 16.3 |
| % annotated by kegg | 42.1 |
| % annotated by eggNOG | 60.44 |
Statistics on 9.9M reference genes
For more information, see:
- Li, J., et al. An integrated catalog of reference genes in the human gut microbiome. Nature biotechnology 2014;32(8):834-841. doi:10.1038/nbt.2942
- Arumugam, M., et al. Supporting data for the paper: "An integrated catalog of reference genes in the human gut microbiome". GigaScience Database 2014 doi:10.5524/100064
- Integrated reference catalog website: http://meta.genomics.cn/metagene/meta/home
Public metagenomes used:
| Reference | giga_bases | sequencing_runs | samples | subjects |
|---|---|---|---|---|
| Human Microbiome Project Consortium. The framework for human microbiome research. Nature 2012;486(7402):215-221. doi:10.1038/nature11209 | 3771.27 | 1576 | 337 | 180 |
| Le Chatelier, E., et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013;500(7464):541-546. doi:10.1038/nature12506 | 1641.29 | 595 | 292 | 292 |
| Li, J., et al. An integrated catalog of reference genes in the human gut microbiome. Nature biotechnology 2014;32(8):834-841. doi:10.1038/nbt.2942 | 1641.29 | 595 | 292 | 292 |
| Nielsen, H.B., et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nature biotechnology 2014;32(8):822-828. doi:10.1038/nbt.2939 | 1455.11 | 1704 | 396 | 318 |
| Zhang, X., et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature Medicine 2015;21(8):895-905. doi:10.1038/nm.3914 | 1454.22 | 232 | 232 | 232 |
| Qin, J., et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490(7418):55-60. doi:10.1038/nature11450 | 1295.18 | 365 | 365 | 365 |
| Qin, N., et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014;513(7516):59-64. doi:10.1038/nature13568 | 1207.97 | 314 | 237 | 237 |
| Feng, Q., et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528. doi:10.1038/ncomms7528 | 778.91 | 312 | 156 | 156 |
| Karlsson, F.H., et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013;498(7452):99-103. doi:10.1038/nature12198 | 460.08 | 147 | 145 | 145 |
| Qin, J., et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464(7285):59-65. doi:10.1038/nature08821 | 402.17 | 264 | 124 | 124 |
| Total | 13937.8 | 6088 | 2271 | 1962 |
Statistics on human gut metagenomes used by MetaQuery
All metagenomes were downloaded from the NCBI Sequence Read Archive.
Datasets were identified using the aid of SRAdb (doi:10.1186/1471-2105-14-19)
FASTQC was used to filter low-quality metagenomes with: read-length <50-bp, average read-quality <20, or >2% of reads with adaptor contamination.